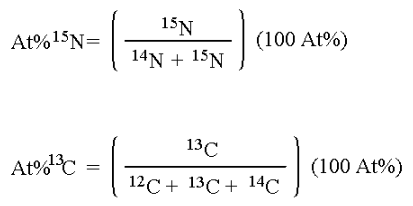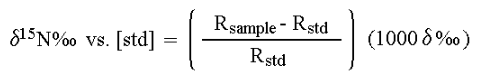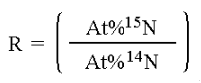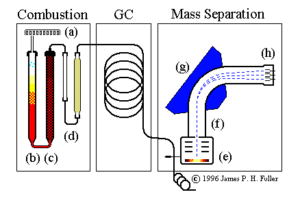
The mass analysis train operated by this laboratory includes the components shown above. These are discussed in the following text.
Combustion subsystem:
- sample dropper (a)
- combustion column (b)
- reduction column (c)
- gas traps (water and optionally CO2) (d)
Gas chromatograph subsystem:
Mass separation subsystem:
- ion beam source (e)
- flight tube (f)
- magnetic beam deflector (g)
- signal detectors (h)
A sample’s isotopic composition is measured by determining the ratios of the stable isotope masses being examined – in the case of this laboratory, 15N/14N or 13C/12C as appropriate. These ratios are measured on an isotope ratio mass spectrometer, a device that separates ions of the element of interest on the basis of their differing mass/charge ratio (m/z).
Combustion
Sample preparation consists of converting solid or liquid material to a gas and isolating the particular gas that must be analyzed (N2 or CO2.) In the machine diagramed at the top of this page a rotating multiplace sample dropper (a) delivers one sample at a time into the top of a quartz combustion tube (b). This tube contains granulated chromium III oxide combustion catalist and is held at 1200 degrees C. A pulse of pure O2 is admitted with each sample. All combustible materials in the sample are flash-burned and the resulting gaseous combustion products are swept out the bottom of the tube by a constant stream of nonreactive helium carrier gas.
All carbon in tha sample is converted by combustion to CO2. Nitrogen-bearing combustion products include N2 gas and various oxides of nitrogen NOx. The gasified sample passes from the combustion tube into another furnace (c) containing fine copper granules at 650 degrees C. In this furnace all molecules of NOx give up their oxygen to the hot copper and emerge as pure N2.
Water vapor from the sample is removed by a gas trap (d) containing magnesium perchlorate. If the samples are being analyzed for nitrogen isotopes only, CO2 is removed by a second gas trap containing a CO2 scrubber (sodium hydroxide on silicate carrier granules).
GC Peak Separation
The clean sample gases now pass through a gas chromatograph column (shown in the diagram block labelled GC) to separate the N2 and CO2 and permit these to reach the mass spectrometer at different times. N2 elutes from the GC column first, then CO2.
Mass Separation
A needle-type splitter valve admits a small portion of the pure gases (~1%) into one end of a highly evacutaed flight tube. A hot filament in the ion source (e) generates a beam of electrons; molecules of N2 or CO2 are ionized by the impact of these electrons. These ions are collimated into a focused beam and accelerated into the flight tube (f). The ion beam enters a strong magnetic field created by an electromagnet (g) which performs the actual mass separation. Ions in the field are deflected into circular paths whose radii are proportional to their masses. Each ion is subject to a centripetal force.
HeV
where
H = magnetic field strength
e = ion charge
V = velocity.
Counteracting this is the ion’s centrifugal force
mV^2/r
where
m = mass
V = velocity
r = radius of curvature.
Solving, we obtain the radius r of each ion’s path:
mV^2r = HeV
r = mV / eH
Or, to paraphrase in English, light ions are deflected more and heavier ones less. The result is that the ion beam is divided into its component masses.
The separated masses finally reach collectors (Faraday cups) at (h), placed so as to receive masses 28, 29, and 30 for dinitrogen or 44, 45, and 46 for carbon dioxide. Here the impact of the ions is translated into a recordable signal that is collected for data processing and analysis. The absolute levels of the signals depend on the amount of gas which is admitted into the ion source. The critical value is the ratio of the signals falling into the different collector cups.
Standard materials and calibrants
In stable isotope analysis, calibration is made against reference materials chosen to have an isotopic abundance and total-element composition (nitrogen or carbon content and carbon-to-nitrogen ratio) similar to the expected values range for the samples. It has been found that measuring the absolute isotopic composition of a sample is not usually as reliable or convenient as measuring isotopic differences between a sample and an appropriate standard. This technique of differential comparison of sample and standard provides high precision and repeatability over both short-term and long-term periods and permits determination of very small differences in the isotopic composition of two samples. To evaluate very small differences in isotopic composition between samples and standards, the isotope ratios of both are determined by mass spectrometry. For example, in 13C analysis the sample is converted to CO2 gas before analysis. Various combinations of the isotopes of C and O can create molecules of the masses shown below but, as will be evident from summing the percentages of the total CO2, only the mass ratios 45/44 and 46/44 need be determined. Though absolute mass ratios are measured, the principle interest lies in the difference between the sample and standard ratios.
Sample prep considerations
Biological materials analyzed for stable isotope content include leaves, roots, soil, plasma and other solid and liquid substances. Before samples can be analyzed by they must be converted into the simple gases N2 or CO2. The micro-Dumas combustion elemental analyzer used here as a front end for the mass spectrometer requires some care in preliminary sample preparation (though considerably less than wet-chemistry techniques such as Kjeldahl-Rittenberg.)
1. Solid samples must be oven-dried (80 degrees C, 24 hours). Freeze-drying must be used if the samples contain forms of N such as ammonia that would lost in oven-drying.
2. Dried samples are ground to talcum powder consistency (250 um or less) using a ball mill (e.g. Spex Industries 8000) before being sealed into 5 x 9 mm tin capsules. Thorough sample homogenization in the grinder stage is required, to make certain that the tiny subsample taken for analysis (e.g. ~2 mg for 13C in leaves) is representative of the total sample. Poor precision can often be traced to inadequate grinding that leaves fibrous matter or visible granules in the sample. Wiley mills do not do an acceptable job of grinding for this application. (Nor does that favorite of low-budget improvization, the home coffee mill.)
3. Combustion capsule formation is critical for successful analytical runs. Please refer to the detailed [sample encapsulation] instructions for further information.
4. All mass spectrometers show a confounding effect of sample size on determined isotope ratio. Samples are therefore weighed after grinding to achieve a uniform sample size. (If total C or N values are desired in addition to 13C/15N, these weights are recorded and used in the data analysis.)
For isotopic analysis, sample size and total element content have a major effect on the analysis. The size of the subsample weighed out depends upon the density of the material as well as its N and C content. An ideal soil or plant sample intended for natural abundance isotopic analysis at this lab would contain 200 micrograms total N and 800 micrograms total C. It may not, of course, be possible to provide both these ideal amounts in one sample. The lower limits for reliable day-to-day operation of our instrument at natural abundance isotope levels appear to be ~25 micrograms total N and ~200 micrograms total C. For best precision and accuracy a preliminary analysis for total element content should be performed so that standards may be closely matched to samples.
Another sample-size constraint is related to the absolute amount of material that can be completely combusted in micro-Dumas apparatus. The maximum burnable total C content is around 2500 micrograms (e.g., for NBS 1572 citrus leaf standard, which is 43.27% C, this works out to a maximum sample size of 5.8 mg of ground leaf.) If, for example, a given sample is so poor in N that it must contain > 2500 ug C in order to achieve 25 ug N then it is essentially not analyzable for 15N. In addition, for element-poor soil samples the sheer bulk of the sample becomes significant. Soil samples of over 50 mg are very difficult to analyze due to rapid ash buildup in the furnace.
5. Nitrogen diffusion samples frequently fall at the lower end of the acceptable range of total N content. There is a small-sample mode available for samples containing less than 50 ug total N and no C. In this mode the oxygen pulse added to improve combustion, which as mentioned earlier contains a trace N impurity, is injected between samples rather than with them. Enough oxygen for a small sample is retained by the combustion catalyst, and this retained oxygen is of course free of the N2 impurity. Small-sample mode removes the need for baseline blanks and the variability these blanks add to the analytical process. However, in order to use this mode of operation we must know ahead of time that a given sample set will consist exclusively of low-N, no-C samples.
6. Liquid samples may be analyzed either by freeze-drying directly into a tin capsule or pipetting onto an inert absorbant substrate.